Фазовым анализом называется установление числа фаз в исследуемом образце, их идентификация (качественный анализ) и определение количественного содержания фаз (количественный анализ).
Рентгенографический анализ поликристаллов является одним из основных методов исследования состава и структуры твердотельных соединений. В ряде случаев он дает уникальную информацию о фазовом составе и строении веществ, которая не может быть получена с помощью других аналитических методов.
Каковы достоинства рентгенографического метода фазового анализа? В отличие от рентгеноспектрального анализа, ИК-спектроскопии, Оже-спектроскопии и т.п., которые позволяют установить лишь элементный состав исследуемых веществ, рентгеноструктурный анализ дает возможность определить, из каких фаз состоит образец. Более того, одно и то же вещество может находиться в различных кристаллических модификациях, что также устанавливается по рентгенограмме.
КАЧЕСТВЕННЫЙ ФАЗОВЫЙ АНАЛИЗ
Качественный фазовый анализ базируется на точном определении положения и интенсивности линий дифракционных спектров.
Различные фазы в кристаллах обладают не только разными свойствами, но и разной кристаллической структурой. От каждой фазы на рентгенограмме возникает свой набор интерференционных линий. Относительные интенсивности и определенные по рентгенограмме межплоскостные расстояния (d) называют рентгеновской характеристикой вещества. Самые сильные линии данного вещества называют реперными. По таким линиям обычно и выявляют фазы. Рентгенограммы многофазной системы представляют собой результат наложения рентгенограмм отдельных фаз, интенсивности которых пропорциональны количеству фазы в системе. Минимальное количество вещества, при котором еще заметны реперные линии, определяет чувствительность фазового анализа.
Чувствительность метода зависит от интенсивности интерференционных линий, которые дает на рентгенограмме исследуемое вещество. Интенсивность линий для порошкового образца определяется произведением ряда факторов:
Ihkl = A(θ,μ)n2λ3L(θ)P(θ)F2(hkl)e-2Mp (1),
где A(θ,μ) – множитель поглощения, зависящий от брэгговского угла θ и линейного коэффициента поглощения μ, который зависит от атомного номера вещества и длины волны рентгеновского излучения λ; n – число элементарных ячеек в единице объема; L(θ) – множитель Лоренца или фактор интегральности, появляющийся при переходе от максимальной интенсивности рассеяния Imax= |F2(hkl)|Φ(Ψ1Ψ2Ψ3); Φ(Ψ1Ψ2Ψ3) – интерференционная функция Лауэ) к интегральной для случая, когда падающий и рассеянный лучи лежат в плоскости, перпендикулярной оси вращения кристалла. Множитель, зависящий от угла поворота кристалла, – фактор Лоренца – в случае поликристалла равен (sin2θcosθ)-1. P(θ)– поляризационный множитель, зависящий от геометрии съемки; F2(hkl) – структурный множитель; e-2M – температурный множитель; p – фактор повторяемости, зависящий от симметрии кристалла.
Из формулы (1) видно, что чувствительность метода зависит от соотношения коэффициентов поглощения определяемой фазы и всей смеси. Фаза с большим μ (т.е. фаза, состоящая из тяжелых элементов с большой рассеивающей способностью) в смеси со слабопоглощающими фазами выявляется при малых ее количествах. При сравнении чувствительности метода для двух фаз с одинаковой решеткой, например кубической, следует учитывать значение структурного фактора. Так, для ОЦК-кристалла он равен 4f2, а для ГЦК-кристалла 16f2, где f2– атомный множитель. Следовательно, одно и то же вещество, имеющее ОЦК- и ГЦК-модификации (например, α-Fe и β-Fe), выявляется в случае ГЦК-структуры при содержании в 4 раза меньшем, чем при наличии ОЦК-решетки.
Фактор повторяемости p определяется симметрией решетки: чем ниже симметрия кристаллической решетки исследуемого вещества, тем больше требуется его для обнаружения. Последнее связано с тем, что понижение симметрии приводит к увеличению количества линий на рентгенограмме. При этом интегральная энергия излучения распределяется на большее число линий и интенсивность каждой из них понижается. Для кристаллов кубической сингонии значения p принимают максимальные значения, поэтому фазы с кубической решеткой дают интенсивные рефлексы. К достоинствам рентгеновского фазового анализа должны быть отнесены также высокая чувствительность и экспрессность метода. Метод не требует большого количества анализируемого вещества; фазовый анализ можно проводить без разрушения образца. Кроме того, метод позволяет оценить количество фаз в смеси.
Чувствительность метода зависит от наличия структурных искажений и дефектов в кристалле и от размера кристаллитов. Эти факторы могут приводить к уширению интерференционных максимумов и, следовательно, к снижению чувствительности метода, поскольку размытые линии выявлять труднее, чем резкие. Чувствительность рентгеновского метода может быть повышена улучшением техники рентгеновской съемки и соответствующей подготовкой исследуемых образцов. Применение монохроматоров уменьшает общий фон рентгенограммы, благодаря чему выявляются слабые линии.
Из-за наличия на рентгенограммах большого числа интерференционных максимумов и возможности наложения максимумов разных фаз рентгенограмму многофазных систем необходимо рентгенографировать в длинноволновом излучении. При работе фотометодом рекомендуется использовать селективно поглощающие фильтры. Надежность идентификации повышается с ростом числа отражений от исследуемой фазы, поэтому следует использовать камеры с большим диаметром (например, РКУ-114). Закладку пленки предпочтительнее осуществлять асимметричным способом.
При рентгенографировании объектов с размером кристалликов более 10-4 – 10-3 см образец необходимо вращать. При этом увеличивается количество зерен вещества, участвующих в отражении, и интенсивность линий возрастает. Однако при анализе двух- и многофазных систем целесообразно получить рентгенограмму без вращения. В этом случае при разном размере частиц фаз, входящих в смесь, характер линий на рентгенограмме от каждой фазы различен. Этот факт может облегчить разделение рефлексов, принадлежащих каждой фазе.
При съемке крупнокристаллических образцов чувствительность метода снижается из-за экстинкции. В отдельных случаях можно рекомендовать измельчение исследуемых порошков, в если объект рентгенографирования – монолитный образец, то наклеп поверхностного слоя следует удалить электрополировкой с последующим отжигом. Однако к механической обработке изучаемых объектов следует подходить очень осторожно, так как при этом можно существенно изменить состояние образца и даже фазового состава смеси.
Применение дифрактометров со сцинтилляционной регистрацией интерференционных максимумов (установки ДРОН-2.0, ДРОН-3.0, ДРОН-4.0) и фокусировкой по Брэггу-Брентано приводит к повышению чувствительности фазового анализа. При такой фокусировке диффузно рассеянные лучи не фокусируются и поэтому уровень фона значительно ниже по сравнению с фотографическим методом регистрации.
Уровень фона на дифрактограмме или рентгенограмме можно уменьшить оптимальным подбором напряжения на рентгеновской трубке. Оно должно составлять 3-4 потенциала ионизации. Например, для трубок с Cu-анодом оптимальное напряжение составляет 25-30 кВ.
Требование обеспечения высокой точности определения интенсивности и низкого порога обнаружения линий, с одной стороны, и прецизионности и экспрессности определения межплоскостных расстояний с другой, приводят к необходимости нахождения компромиссных решений.
Чтобы уменьшить статистические флуктуации счета при регистрации, интенсивности дифракционных рефлексов, необходимо пользоваться широкими пучками, задавать большие постоянные времени и малые скорости сканирования.
Для точного определения 2θ и достижения высокого разрешения линий при больших скоростях сканирования необходимы узкие щели и малые постоянные времени.
Наибольшее действие на смещение и искажение профиля линии оказывают горизонтальная и вертикальная расходимости первичного пучка рентгеновских лучей. Применительно к отечественным рентгеновским трубкам при угле выхода первичного пучка в 6° оптимальная ширина приемной щели составляет ≈ 0,25 мм.
В табл. 1 приведены оптимальные условия регистрации дифрактограмм от поликристаллических образцов для фазового анализа.
Если исключить все систематические ошибки, кроме ошибки промера рентгенограммы, можно рассчитать ошибку в величине межплоскостного расстояния между линиями на рентгенограмме.
Расчет показывает, что при обычной точности промера пленки ±0,1-0,2 мм (или ±Δθ 0,05-0,1°) при съемке в железном излучении, межплоскостное расстояние в 3 Å определяется с точностью до ≈0,01 Å, а межплоскостное расстояние в 1 Å – с точностью до ≈0.001 Å.
Таблица 2.
Оптимальные условия для регистрации дифракционных спектров
Параметры прибора |
ДРОН-3 |
ДРОН-2 |
Рентгеновская трубка БСВ |
23 и 24 |
22 и 23 |
| Анод | Медь | Медь |
| Угол выхода первичного пучка, град. | 6 |
6 |
| Напряжение, кВ | 40 |
40 |
| Ток, мА | 30 |
25 - 30 |
| Ограничивающая щель, мм | ||
| ширина | 2 - 4 |
2 - 4 |
| высота | 8 - 10 |
8 - 10 |
| Приемная щель, мм | ||
| ширина | 0.25 |
0.1 - 0,25 |
| высота | 8 - 10 |
8 - 10 |
Скорость движения счетчика, °/мин |
2 - 1 |
2 - 1 |
Скорость движения ленты, мм/ч |
600 |
600 |
Постоянная времени интенсиметра, с |
2 - 5 |
2 - 4 |
МЕТОДИКА КАЧЕСТВЕННОГО ФАЗОВОГО АНАЛИЗА
Если образец однофазный, то определение фазы представляется простой задачей. Из экспериментального набора межплоскостных расстояний выбирают 3 или 5 самых интенсивных рефлексов. Межплоскостные расстояния отобранных линий сопоставляются с величинами, имеющимися в справочных таблицах. После предварительной идентификации исследуемого вещества весь набор межплоскостных расстояний (d/n) последнего следует сравнить с полной таблицей межплоскостных расстояний и интенсивностей стандарта. Критерием правильности определения вещества будет полное совпадение (в пределах погрешности эксперимента) всех межплоскостных расстояний и отвечающих им интенсивностей рефлексов.
Сложность определения фазового состава возрастает с увеличением числа фаз, из которых состоит исследуемый образец. Для более успешного проведения фазового анализа необходимо привлекать данные химического и спектрального анализов по элементному составу образца.
Для выполнения качественного фазового анализа часто пользуются картотекой ASTM (American Society for Testing Materials), содержащей более 25000 карточек. Карточка разделена на несколько полей, содержащих следующую информацию о веществе: значения межплоскостных расстояний d/n, соответствующие трем сильнейшим линиям рентгендифракционного спектра, информацию об условиях получения спектра (источник рентгеновского излучения, длина волны используемого излучения, диаметр рентгеновской камеры или тип установки, метод получения и т.п.), информацию о структуре соединения – сингония, пространственная группа симметрии, значения периодов кристаллической решетки, угловые параметры элементарной ячейки α, β, γ; число атомов на элементарную ячейку, рентгеновскую плотность, структурную формулу соединения, полный рентгеновский спектр – d/n , I/I1 , (hkl) и номер карточки. I/I1 – отношение интенсивности линии спектра к наиболее сильной линии (относительная интенсивность), (hkl) – индексы интерференции. Образец карточки приведен на рис.1.
Образец карточки ASTM
| d | 2.67 | 2.48 | 1.67 | 3.63 | Cr2O2 Chromium (III) Oxide |
|||||
| I/I1 | 100 | 96 | 90 | 74 | ||||||
| Rad. Cu λ 1.5405 Filter | d, Å | I/I1 | hkl | d, Å | I/I1 | hkl | ||||
Dia. Cut off Coll I/I1 d corr. abs Ref. Swanson et al., NBS Circular 539 Vol. V (1955) |
3.633 2.666 2.480 2.264 |
74 100 96 12 |
012 104 110 006 |
0.9370 .8957 .8883 .8658 |
12 14 7 23 |
410 1.3.10 3.0.12 416 |
||||
Sys. Hexagonal S. G. an 4.954 bnco 13.584 A C α β γ Z6 Ref. Ibid. |
2.176 2.048 1.815 6 1.672 1.579 |
38 9 39 9 0 13 |
113 202 024 116 122 |
.8425 .8331 .8263 .7977 |
8 11 9 15 |
4.0.10 1.0.16 330 3.2.10 |
||||
εα nωβ εγ Sign 2V D Ref. |
1.465 1.431 4 1.296 1 |
25 40 20 |
214 300 1.0.10 |
|||||||
Sample from Jonson, Matthey and Co. Ltd. Spect anal.: <0.001% Ca, Mg; <0.0001% Si, Cu. X-ray pattern at 26°C. Fe2O3 structure type. |
1.2398 1.2101 1.173 1.148 1.1239 |
17 7 14 10 10 |
220 306 128,312 0.2.10 134 |
|||||||
Replaces 1—1294, 2—1362, 3—1124, 4—0765 |
1.087 1.042 0.9462 |
17 16 13 |
226 2.1.10 324 |
|||||||
Рис. 6.
Методику рентгенофазового анализа проводят с применением различных указателей. О них можно прочитать в специальной литературе. Однако проводить такой анализ без применения вычислительной техники довольно сложно. Для облегчения процедур идентификации существуют различные автоматизированные системы. Одной из таких программ является программа БАЗА.
Программа работает в 14 основных режимах, определяемых: 1) способом ввода исходных данных (d, I) – из архива, вручную, путем моделирования; 2) использованием в процессе поиска химических данных (сведения об элементном составе образца); 3) способом сканирования Базы Данных (БД) (просмотр всей БД или сопоставление исходной рентгенограммы со списком задаваемых пользователем вероятных фаз).
Инструкция по проведению фазового анализа
с помощью программы БАЗА
В программе БАЗА находим подпрограмму IDENT.EXE и выполняем ее ввод. Далее:
- выбираем нужную БД (FREVEL(минералы), HIGHTEMP(высокотемпературная), UNORGAN(неорганическая)), выделяем ее и осуществляем ввод
- выбираем один из трех режимов ввода исходной рентгенограммы ( а) из архива, б) ручной ввод)
- моделирование
При ручном вводе появляются запросы относительно способа ввода максимумов отражений – в шкале d или в шкале 2θ. В последнем случае необходимо задать длину волны используемого излучения λ. Значения d задаются в Å, (2θ – в градусах), значения относительных интенсивностей I – в целых числах в стобалльной шкале. Пара чисел d, I задается через запятую, например: 1.219,30. Исходная рентгенограмма может содержать до ста линий. Созданная рентгенограмма выводится на экран дисплея, в виде столбцов межплоскостных расстояний и значений относительных интенсивностей. Клавиши "D/U" позволяют корректировать рентгенограмму (удалять или восстанавливать линию). По клавише "С" осуществляется продолжение работы. После корректировки рентгенограммы появляется команда "V" – (saVe) – сохранить, т.е. записать в архив с присвоением нужного имени. Программа предполагает задание возможного элементного состава. По клавише "С" на экране высвечивается периодическая система Менделеева. Нужный химический элемент на ходится с помощью клавиш "Up","Down" и вводится командой "Enter". Далее на экране появляются запросы по способу поиска:
- по БД
- путем сопоставления с рядом задаваемых пользователем вероятных фаз.
В нашей работе будем пользоваться первым способом введения. Необходимо ввести параметры поиска: граничные критерии соответствия, величины окна отсечки интенсивности и окна углов. Рекомендуемые значения R1, R2 – 40-70, R3, R4 – 60-80, величины окна отсечки – 0.1-0.4. Например: 60; 70; 80; 80; 1; 0.2. Во всех случаях критерии R1, R2, R3, R4 оценивают численно степень соответствия рентгенограммы стандарта или элементного состава стандарта предъявляемым исходным данным. После ввода клавиши "С" начинается процесс идентификации. На экран выводится таблица с результатами идентификации. Для примера приведена таблица 2 идентификации результатов поиска фаз в неизвестной смеси, содержащей атомы К, Na и Сl.
В первом столбике таблицы занесен порядковый номер предполагаемой фазы, во втором – номер карточки JCPDS, в третьем – полная информативность рентгеновского стандарта и информативность части рентгенограммы, попадающей в заданные границы. Всем химическим элементам присвоены веса в соответствии с их информативностью. Минимальный вес, равный 20, у кислорода и водорода. Максимальный вес, равный 125, у наиболее редких элементов, у лантоноидов и актиноидов. В четвертом столбце приведено отношение числа линий в рентгенограмме образца, совпавшее с числом линий в рентгенограмме стандарта (в границах рентгенограммы образца). В следующих столбцах даны значения критериев R1, R2, R3, R4. Они показывают соответствие между рентгенограммой и выбранными стандартами на четырех стадиях идентификации. На 1-й и 2-й стадиях вычисляются грубые критерии соответствия R1 и R2 между сравниваемыми рентгенограммами. На 3-й и 4-й стадиях вычисляют более точные критерии R3 и R4 по 8 и 40 сильнейшим линиям соответственно, с учетом как положения, так и значений интенсивности образца и стандарта. Все критерии нормированы на 100. В последнем столбце приведена химическая формула стандарта. Из таблицы результатов идентификации поиска (табл. 3 ) выбирают не более 4 стандартов и выделяют их с помощью клавиши "Enter".
Таблица 3
Результаты идентификации
N
|
JCPDS
|
Inform Uf/Us
|
Nc/Ns
|
Factors |
Chemical Formula |
|||
| R1 | R2 | R3 | R4 | |||||
1 |
5-628 |
34/41 |
14/14 |
80 |
90 |
99 |
99 |
NaCl |
2 |
18-225 |
30/40 |
7/9 |
60 |
78 |
98 |
98 |
(Be Pd) |
3 |
10-220 |
29/33 |
10/11 |
80 |
80 |
97 |
97 |
SiCl4 |
4 |
4-587 |
32/36 |
12/12 |
70 |
100 |
97 |
97 |
KCl |
5 |
12-149 |
67/93 |
12/15 |
60 |
62 |
96 |
96 |
Bi4MoO9 |
6 |
15-898 |
50/66 |
12/14 |
60 |
82 |
96 |
96 |
(PuP) |
7 |
27-152 |
31/34 |
12/16 |
70 |
70 |
96 |
96 |
(Cu Zn2 Ga Si4) |
8 |
11-616 |
48/48 |
8/10 |
60 |
70 |
95 |
95 |
Fe3Si |
Далее предусмотрена возможность вывода сравнительных таблиц, содержащих рентгеновские характеристики исследуемого образца и стандартов, как на экран, так и на печать, и вывода штрихдиаграмм.
В табл. 4 приведены результаты качественного фазового анализа. Как видно из табл. 4 , набор межплоскостных расстояний и значения относительных интенсивностей в исследуемой смеси хорошо соответствуют таковым значениям в фазах КСl и NaCl;
Окончательное решение о составе образца пользователь принимает на основе анализа табличного и графического соответствия рентгенограмм образца и ряда интересующих фаз. Следует учитывать, что фазы малой концентрации могут быть отсечены на какой-либо стадии поиска, если заданы слишком высокие значения граничных критериев соответствия. В этом случае надо изменить эти граничные значения в сторону уменьшения и повторить поиск вновь.
Соответствие между экспериментальной рентгенограммой и стандартами на заключительной четвертой стадии поиска (табл. 4) составляет 97% и 99% соответственно.На рис. 7 приведена штрихдиаграмма образца в сопоставлении со штрихдиаграммами выбранных стандартов.
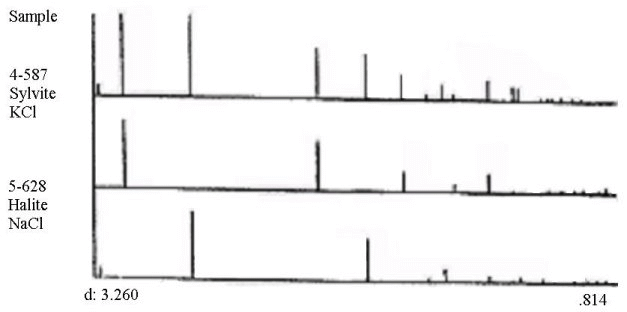
Как видно из табл. 4 , исследуемый образец представлен двумя фазами: КСl и NaCl. Идентификация произведена по 8 неперекрывающимся отражениям от КС1 и 10 неперекрывающимся отражениям от NaCl .
Линии исходной рентгенограммы, непроидентифицированные в рамках выбранной совокупности фаз, помечены звездочкой. В нижней части табл. 3 (под чертой) для каждого стандарта перечислены линии, не входящие в область задания исходной рентгенограммы.
После предварительной идентификации исследуемого объекта, можно:
- изменить параметры поиска стандартных фаз;
- перейти на ввод новой исходной рентгенограммы;
- закончить работу.
С помощью пакета программ "БАЗА" можно обрабатывать экспериментальные кривые рассеяния, полученные на автоматизированной установке типа ДРОН-4. В этом случае создается массив интенсивностей и углов рассеяния.
Таблица 4
Результаты качественного фазового анализа
Sample |
4-587 97 * Silvite KCl |
5-628 99* Halite NaCl |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Данный массив имеет расширение r01. Для дальнейшей обработки экспериментальных данных с помощью подпрограммы "DRNTOMID" происходит перевод массива из кодов ЭВМ рентгеновской установки в коды персонального ЭВМ. Вследствие этого получаем файл с расширением mid. Например, drntomid имя файла01.г01 имя файла0l.mid. Например: drntomid oct01.r0l oct01.mid. С помощью подпрограммы "SPECTR1" выделяем максимумы интенсивности рассеяния на дифракционной картине и рассчитываем значения межплоскостных расстояний и относительные интенсивности. Результатом проделанной работы будет создание файла с расширением std, который обрабатывается по вышеописанной методике с помощью программы "IDENT".
Порядок выполнения работы
- Получить рентгенограмму от исследуемого образца дифрактометрическим или фотографическим методами.
- Определить положения брэгговских максимумов и оценить относительные интенсивности вручную или с помощью подпрограмм "DRNTOMID" и "SPECTR1", если распределение интенсивности рассеяния получено при помощи автоматической установки ДРОН-4.0.
- Путем сопоставления экспериментально полученных значений d/n и Iотн с табличными значениями межплоскостных расстояний и относительных интенсивностей рассеяния определить фазовый состав образца.
- С помощью подпрограммы "IDENT" определить фазовый состав образца и сравнить результаты ручной и машинной обработки данных.
- Вывести на печать результаты выполненной работы:
- штрихдиаграммы исследуемого образца и выбранных стандартов,
- таблицы межплоскостных расстояний и относительных интенсивностей для образца и подобранных стандартов соответственно.
- Проанализировать полученные результаты, сопоставить их с данными химического анализа.
КОНТРОЛЬНЫЕ ВОПРОСЫ
- Что называют фазовым анализом?
- Обосновать возможность применения рентгеновских лучей к исследованию фазового состава смесей.
- В чем заключается преимущество рентгенофазового анализа перед другими (какими) методами?
- Какова чувствительность рентгеновского фазового анализа?
- Какие требования предъявляются к выбору излучения, к образцу и условиям регистрации дифрагированного излучения?
КОЛИЧЕСТВЕННЫЙ ФАЗОВЫЙ АНАЛИЗ
Количественный фазовый анализ является вторым этапом, когда качественный фазовый состав известен.
Количественный рентгеновский фазовый анализ основан на зависимости интенсивности дифракционного отражения от содержания соответствующей фазы, распределённой в исследуемом многофазном поликристаллическом образце. При одинаковом содержании определяемой фазы интенсивность дифракционного отражения будет меняться в зависимости от величины среднего коэффициента поглощения рентгеновских лучей в образце. Необходимо либо найти эту зависимость и определить коэффициент поглощения образца, либо использовать методы, позволяющие устранить влияние фактора поглощения.
Рассмотрим съемку "на отражение" поликристаллического образца, содержащего n фаз (рис. 8 ).
Схема съемки на отражение для плоского образца "IDENT"
Рис. 8
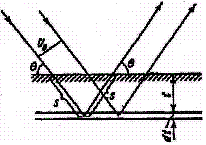
На образец под углом θ падает первичный пучок с сечением U0. Объем отражающего элементарного слоя dV на глубине t равен ![]() . Обозначим объемное содержание в образце определяемой фазы через ƒi, тогда интенсивность дифракционного отражения, создаваемого этой фазой, находящейся в объеме dV, составит:
. Обозначим объемное содержание в образце определяемой фазы через ƒi, тогда интенсивность дифракционного отражения, создаваемого этой фазой, находящейся в объеме dV, составит:
dIi = (Ii)0ƒiU0e-2μsdS (1),
где (Ii)0 – интенсивность дифракционного отражения образца, состоящего только из i-й фазы без учета поглощения. Считая исследуемый образец бесконечно толстым, получаем значение интенсивности рассеяния
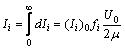 (2),
(2),
Обозначая ![]() , получим
, получим
![]() (3).
(3).
Заменим объемные проценты ƒiна весовые xi. Обозначим через wi, pi, νi соответственно вес, плотность и объем i-ой фазы. Тогда вес образца ![]() , объем образца
, объем образца ![]() , тогда
, тогда  .
.
Разделив числитель и знаменатель на W , получим
![]() (4)
(4)
Массовый коэффициент поглощения выражается через массовые коэффициенты поглощения фаз:
![]() ;
;

Из двух последних формул получим выражение для линейного коэффициента поглощения образца μ через массовые коэффициенты поглощения составляющих фаз – μ*
 (5)
(5)
Подставляя (4) и (5) в (3), получим
![]() (6)
(6)
Если образец содержит две фазы, то
![]() (7).
(7).
В принципе к формуле (7) можно свести определение содержания фаз и в том случае, когда образец n-фазный, если коэффициент поглощения матрицы заменить коэффициентом поглощения (n-1)фазы.
Рассмотрим различные варианты количественного фазового анализа, определяемые соотношениями между коэффициентами поглощения фаз и образца в целом.
Средний коэффициент поглощения образца не меняется. Тогда зависимость (6) трансформируется к прямо пропорциональной зависимости Ii = kixi. Коэффициент ki определяется посредством съемки эталона – образца с известным содержанием исследуемой фазы и с неизменным коэффициентом поглощения. Можно построить линейный график по нескольким точкам, полученным путем съемки нескольких эталонных образцов. Постоянство коэффициента поглощения будем иметь место, если образец представляет собой смесь полиморфных модификаций.
Когда коэффициенты поглощения фаз сильно отличаются, интенсивность рассеяния уже не будет пропорциональна содержанию фазы, т.к. она будет зависеть еще и от соотношения коэффициентов поглощения исследуемых фаз (7). Вычислив μ1* и μ2* на основании табличных данных, можно определить постоянный коэффициент в выражении (7) путем съемки образца с известным фазовым составом. Например, из рентгенограммы образца, содержащего 100% определяемой фазы, имеем
![]() (8)
(8)
Разделив выражение (7) на выражение (8) , получим
 (9)
(9)
Сняв второй эталонный образец, можно определить ![]() .
.
Теоретически построенные кривые по формуле (7) приведены на рис. 9 .
Зависимость формы градуировочного графика
от отношения коэффициентов поглощения
определяемой фазы μ1 и вмещающей среды μ2

Содержание определяемой фазы в весовых частях
Рис. 9.
μ1/μ2 = 4 - 1; 2,5 - 2; 1 - 3; 0,5 - 4; 0,25 - 5.
Как видно из рис. 9 , отклонение от прямолинейной зависимости будет тем больше, чем больше разница в коэффициентах поглощения фазμ1и μ2. Полученный график используют далее для нахождения данной фазы в любом другом образце неизвестного состава.
Эту графическую зависимость можно построить экспериментально по ряду образцов с известным содержанием фаз.
Другой вариант анализа двухфазных объектов заключается в определении отношения интенсивностей линий обеих фаз:
![]() (10)
(10)
Чтобы определить содержание неизвестной фазы, необходимо отснять, как минимум, два эталонных образца, одним из которых может быть образец, содержащий 100% определяемой фазы.
Требования к эталону
- Линии эталона не должны совпадать с сильными линиями от исследуемого образца.
- Коэффициент поглощения эталона должен быть приблизительно равен коэффициенту поглощения исследуемой фазы.
- Размер кристаллитов не должен превышать 5-25 мкм. Эталонное вещество перемешивают с порошком исследуемого вещества с помощью механической мешалки в метаноле в течение 10 часов. За это время жидкость испаряется и достигается равномерное распределение компонентов.
Требования к препарату для количественного фазового анализа
Степень измельчения материала играет большую роль при количественных измерениях. Недостаточное измельчение приводит к систематической ошибке из-за микропоглощения и экстинкции. Случайная ошибка, вызываемая флуктуациями числа отражающих кристаллитов, резко возрастает при увеличении средних размеров кристаллитов. При значительном измельчении образца интенсивность падает за счет искажений кристаллической решетки на поверхности зерен. На основе экспериментальных данных сделан вывод, что средний размер кристаллитов должен находиться в пределах 5-40 мкм. Крупнокристаллические образцы должны измельчаться до прохождения сита – 104 отверстий на 1 см2. Для уменьшения ошибки, вследствие флуктуации числа отражающих кристаллитов, рентгенографирование необходимо проводить либо с вращением образца в своей плоскости, либо с колебаниями около главной оси гониометра. Образование в объекте исследования преимущественной ориентировки изменяет величину интенсивности дифракционного отражения. Для уменьшения вероятности образования текстуры рекомендуется следующая методика приготовления образца: размеры кристаллитов должны быть в пределах указанных, а глубина держателя образцов – большой. Преимущественная ориентировка кристаллитов образуется при выравнивании поверхности образца уплотнением, поэтому такой способ формирования поверхности следует отвергнуть, а удаление лишнего материала из кюветы производить при помощи лезвия. Поскольку преимущественная ориентировка возникает в тонком наружном слое, то она опасна для сильно поглощающих образцов. Поэтому рекомендуется рационально разбавлять материал образца слабо поглощающей средой, чтобы отражения происходили не только с поверхности образца, но и от внутренних слоев с хаотично ориентированными кристаллитами.
АНАЛИЗ ПОГРЕШНОСТЕЙ ПРИ ПРОВЕДЕНИИ КОЛИЧЕСТВЕННОГО ФАЗОВОГО АНАЛИЗА
Все погрешности можно условно разделить на три группы:
- Погрешности метода (систематические ошибки).
- Погрешности, связанные с приготовлением образцов (случайные).
- Аппаратурные погрешности (систематические и случайные).
Погрешности метода обусловлены следующими причинами:
- неточным учетом коэффициента поглощения (а)
- некорректным учетом наложения интерференционных линий (б)
- кристаллохимическим различием анализируемых фаз в исследуемых пробах и эталонных смесях, используемых для построения градуировочного графика (в)
- неточным построением градуировочного графика (г)
Из всех перечисленных погрешностей данной группы наиболее существенны первые. Более точный учет коэффициента поглощения обеспечивается в методе внутреннего стандарта. Ошибки, связанные с наложением линий, исключают путем подбора аналитических линий без наложения, либо используя метод, учитывающий наложение. Ошибки в) и г) уменьшают путем рационального подбора эталонных смесей и многократным построением каждой точки градуировочного графика.
Погрешности, связанные с приготовлением образцов, обусловлены:
- отбором пробы – проба должна содержать все фазы
- невоспроизводимостью состояния образца из-за наличия текстуры, микроискажений, зкстинкции и т.д.
- флуктуациями числа кристаллитов – необходимо рентгенографирование образца производить с вращением его вокруг нормали к поверхности
- неравномерностью распределения введенного стандарта в образец
Аппаратурные погрешности связаны со случайными ошибками счета и нестабильностью работы установки. К систематическим погрешностям относится нелинейность интенсиметра. Эти ошибки наиболее существенны при малом количестве анализируемой фазы. Для уменьшения случайных погрешностей необходимо увеличить светосилу прибора. Дрейф установки учитывается периодической съемкой эталона по нескольку раз в день.
ЗАДАНИЕ
Исследовать системы вольфрам-молибден и алюминий-медь с различным процентным содержанием компонентов.
Порядок выполнения работы
- Получить дифрактограммы для чистых компонентов, эталонных и двух смесей неизвестного состава (р и q).
- Измерить интегральные интенсивности для двух-трех линий каждого состава и определить относительные интенсивности.
- Построить графики
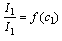 .
. - Используя полученные графики, определить содержание исследуемых компонентов в двух предложенных смесях.
- Построить зависимость
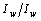 как функцию концентрации для всех смесей.
как функцию концентрации для всех смесей. - Построить эту же зависимость теоретически, используя формулу (7), и сравнить с экспериментально полученной.
КОНТРОЛЬНЫЕ ВОПРОСЫ
- На чем основан количественный фазовый анализ?
- Какие методы количественного рентгенофазового анализа Вы знаете?
- Какой метод использовался в данной работе?
- Какие требования предъявляются к образцам для количественного фазового анализа?
- Достоверность количественного фазового анализа. Основные погрешности метода.
ЛИТЕРАТУРА
- Хейкер Д.М., Зевин Л.С. Рентгеновская дифрактометрия. М.:Физматгиз, 1954. 380 с.
- Васильев Е.К., Нахмансон М.С. Качественный ретгенофазовый анализ. Новосибирск: Наука, 1986. 200 с.
- Нахмансон М.С., Фекличев В.Г. Диагностика состава материалов рентгендифракционными и спектральными методами. Л.: Машиностроение, 1990. 356 с.
- Русаков А.А. Рентгенография металлов. М.: Атомиздат, 1977. 480 с.
- Горелик С.С., Расторгуев Л.Н., Скаков Ю.А. Рентгенографический и электроннооптический анализ. М.: Металлургиздат, 1970. 352 с.
- Миркин Л.И. Справочник по рентгеноструктурному анализу поликристаллов. М.: Физматгиз,1961. 864 с